Testing et scoring du PD-L1

Testing et Scoring du PD-L1
Un biomarqueur, deux méthodes de scoring1
Le PD-L1 est un biomarqueur immunitaire qui peut être exprimé sur diverses cellules telles que les cellules tumorales et cellules immunitaires associées à la tumeur (ex. macrophages et lymphocytes).2-4
- L’expression du PD-L1 est mesurée avec le score de proportion tumorale (TPS) ou le score positif combiné (CPS).1
- Utilisez la méthode d’expression du PD-L1 appropriée pour chaque type de tumeur.1
Méthodes de scoring
Le scoring avec TPS ou CPS dépend du type de la tumeur4
Pour TPS et CPS, utilisez:
- Kit CDx (Agilent) : PD-L1 IHC 22C3 PharmDx (ou test concordant)
- Plateforme : AutoStainer Link 48 (ou plateforme analogue)
- Méthode de coloration : Immunohistochimie4
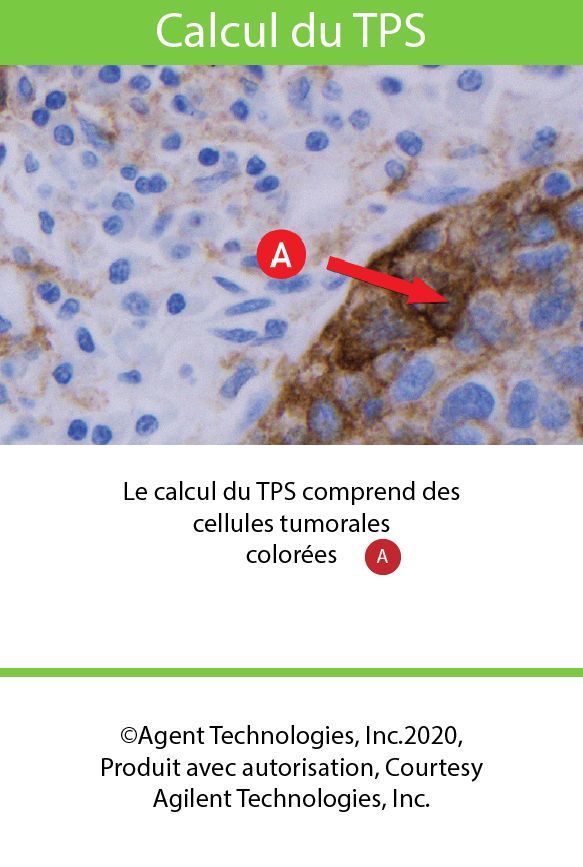
Calcul du TPS3

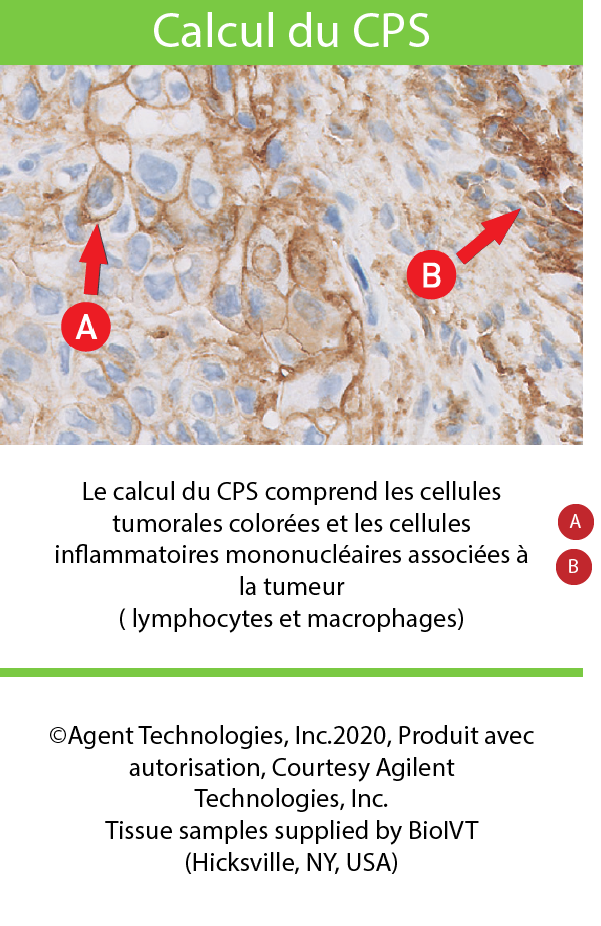
Calcul du CPSa,3

aBien que le résultat du calcul puisse dépasser 100, le score maximal est défini comme CPS 100
Testez avec PD-L1 IHC 22C3 PharmDx ou test concordant pour déterminer le TPS ou le CPS3
Les bases du testing et du scoring
Pour le TPS et le CPS, on évalue des cellules spécifiques3
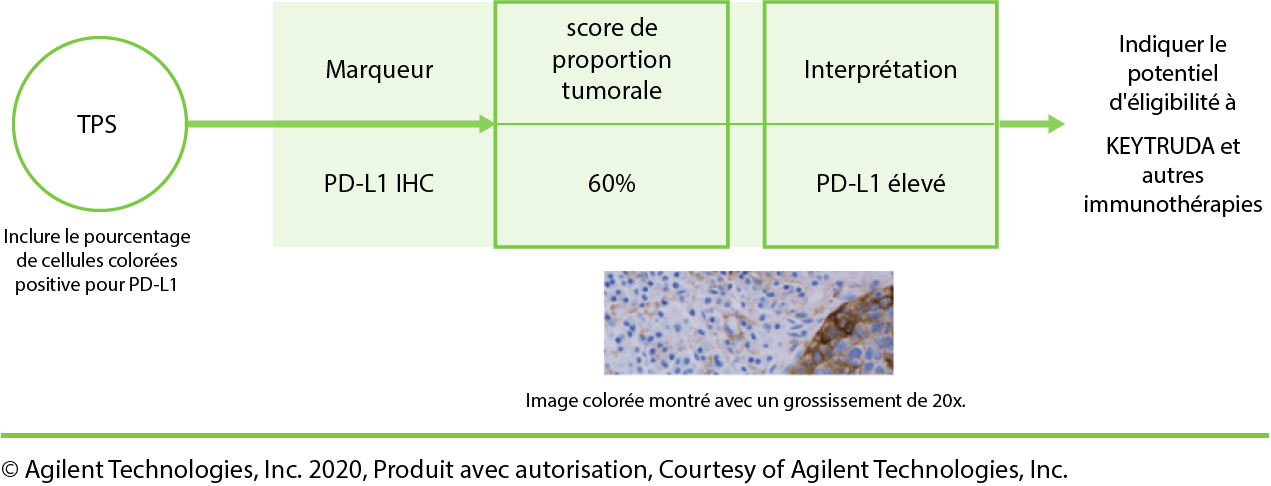
- Le TPS est le pourcentage de cellules tumorales viables présentant une coloration partielle ou complète de la membrane à n’importe quelle intensité.
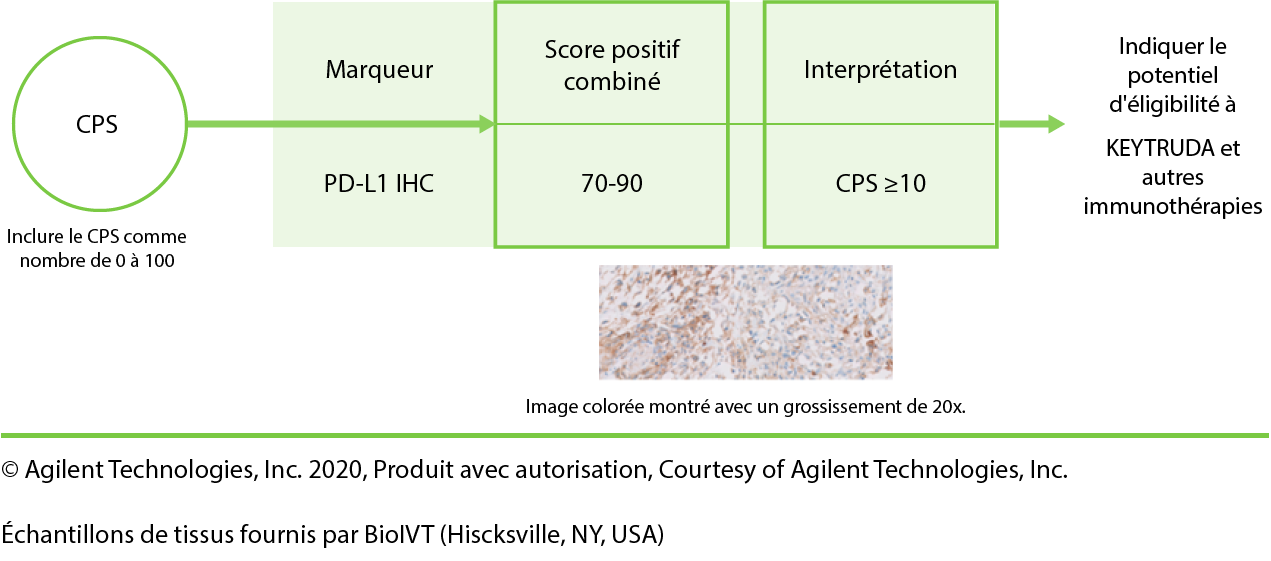
- Le CPS est le nombre de cellules colorées PD-L1 (cellules tumorales, macrophages lymphocytes) par rapport à toutes les cellules tumorales viables.
évaluez des cellules pertinentes lors du calcul du TPS ou CPS du PD-L13
En incluant les calcul TPS et CPS à vos rapports d’anatomopathologie, vous pouvez aider à la prise de décision thérapeutique.3
cut points pour KEYTRUDA® (pembrolizumab)




Reporting
Inclure le score approprié (TPS ou CPS) et l’interprétation pour le type de tumeur4
Rapport du TPS
Rapport du CPS
TPS est rapporté sous forme de pourcentage sur une échelle de 0 % à 100 %.1
Bien que le résultat du calcul CPS puisse dépasser 100, le score maximal est défini comme CPS 1003
Pour optimiser votre rapport, utilisez la bonne méthode du calcul du score de l’expression PD-L1 pour chaque type tumoral1
ALK = kinase lymphome anaplastique; CDx = diagnostic compagnon; CPS = Score positif combiné; EC = cancer de l’œsophage; EGFR = récepteur du facteur de croissance épidermique; GEJ = jonction gastro-oesophagienne; IHC = immunoohistochimie; NSCLC cancer du poumon non à petites cellules; PD-L1 = récepteur 1 de mort cellulaire programmée. 1; TNBC = cancer du sein triple négatif ; TPS = Score de proportion tumorale; uc = Carcinome urothélia
Mentions légales réduites
KEYTRUDA 25 mg/mL solution à diluer pour perfusion. Indications thérapeutiques : Mélanome KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et des adolescents âgés de 12 ans et plus atteints d’un mélanome avancé (non résécable ou métastatique). KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes et des adolescents âgés de 12 ans et plus atteints d’un mélanome de stade IIB, IIC ou III, ayant eu une résection complète Cancer bronchique non à petites cellules (CBNPC) KEYTRUDA est indiqué en monothérapie dans le traitement de première ligne des patients adultes atteints d’un cancer bronchique non à petites cellules métastatique dont les tumeurs expriment PD-L1 avec un score de proportion tumorale (TPS)≥ 50 %, sans mutations tumorales d’EGFR ou d’ALK. KEYTRUDA, en association à une chimiothérapie pemetrexed et sel de platine, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique non-épidermoïde dont les tumeurs ne présentent pas de mutations d’EGFR ou d’ALK. KEYTRUDA, en association au carboplatine et au paclitaxel ou au nab-paclitaxel, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique épidermoïde. KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints de cancer bronchique non à petites cellules localement avancé ou métastatique dont les tumeurs expriment PD-L1 avec un TPS ≥1 %, et ayant reçu au moins une chimiothérapie antérieure. Les patients présentant des mutations tumorales d’EGFR ou d’ALK doivent également avoir reçu une thérapie ciblée avant de recevoir KEYTRUDA. Lymphome de Hodgkin classique (LHc) KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et pédiatriques âgés de 3 ans et plus atteints d’un lymphome de Hodgkin classique en rechute ou réfractaire après échec d’une greffe de cellules souches (GCS) autologue ou après au moins deux lignes de traitement antérieures lorsque la GCS autologue n’est pas une option de traitement. Carcinome urothélial KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome urothélial localement avancé ou métastatique ayant reçu une chimiothérapie antérieure à base de sels de platine.KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome urothélial localement avancé ou métastatique inéligibles à une chimiothérapie à base de cisplatine et dont les tumeurs expriment PD-L1 avec un score positif combiné (CPS) ≥ 10. Carcinome épidermoïde de la tête et du cou (CETEC) KEYTRUDA est indiqué en monothérapie ou en association à une chimiothérapie à base de sels de platine et de 5-fluorouracile (5-FU) dans le traitement de première ligne des patients adultes atteints d’un carcinome épidermoïde de la tête et du cou métastatique ou récidivant non résécable dont les tumeurs expriment PD-L1 avec un CPS ≥ 1. KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome épidermoïde de la tête et du cou récidivant ou métastatique dont les tumeurs expriment PD-L1 avec un TPS ≥ 50 % et en progression pendant ou après une chimiothérapie à base de sels de platine. Carcinome à cellules rénales (CCR) KEYTRUDA, en association à l’axitinib, est indiqué dans le traitement de première ligne des patients adultes atteints d’un carcinome à cellules rénales avancé. Cancers avec instabilité microsatellitaire élevée (MSI-H) ou déficience du système de réparation des mésappariements de l’ADN (dMMR) Cancer colorectal KEYTRUDA est indiqué en monothérapie pour le traitement de première ligne d’un cancer colorectal métastatique MSI-H ou dMMR chez des patients adultes. Cancers non-colorectaux KEYTRUDA est indiqué en monothérapie dans le traitement des tumeurs MSI-H ou dMMR chez les patients adultes atteints de cancer de l’endomètre avancé ou récidivant, dont la maladie progresse pendant ou après un traitement antérieur à base de sels de platine reçu quel que soit le stade et qui ne sont pas éligibles à une chirurgie curative ou à une radiothérapie. Cancer du sein triple négatif (CSTN) KEYTRUDA, en association à une chimiothérapie, est indiqué dans le traitement des patients adultes atteints d’un cancer du sein triple négatif localement récurrent non résécable ou métastatique, dont les tumeurs expriment PD- L1 avec un CPS ≥ 10 et qui n’ont pas reçu de chimiothérapie antérieure pour la maladie métastatique. Cancer de l’endomètre (CE) KEYTRUDA, en association au lenvatinib, est indiqué dans le traitement des patientes adultes atteintes d’un cancer de l’endomètre avancé ou récidivant, dont la maladie progresse pendant ou après un traitement antérieur à base de sels de platine reçu quel que soit le stade et qui ne sont pas éligibles à une chirurgie curative ou à une radiothérapie. Posologie et mode d’administration Le traitement doit être initié et supervisé par des médecins qualifiés et expérimentés dans l’utilisation de traitements anticancéreux. Test PD-L1 Si cela est spécifié dans l’indication, la sélection des patients pour le traitement par KEYTRUDA basée sur l’expression tumorale de PD-L1 doit être confirmée par un test validé Test MSI/MMR Si cela est spécifié dans l’indication, la sélection des patients pour le traitement par KEYTRUDA basée sur le statut tumoral MSI- H/dMMR doit être confirmée par un test validé. Posologie La dose recommandée de KEYTRUDA chez les adultes est soit de 200 mg toutes les 3 semaines, soit de 400 mg toutes les 6 semaines, administrée en perfusion intraveineuse pendant 30 minutes. La dose recommandée de KEYTRUDA en monothérapie chez les patients pédiatriques âgés de 3 ans et plus atteints d’un LHc ou chez les patients âgés de 12 ans et plus atteints d’un mélanome est de 2 mg/kg de poids corporel (jusqu’à un maximum de 200 mg) toutes les 3 semaines, administrée en perfusion intraveineuse pendant 30 minutes. Pour une utilisation en association, voir le Résumé des Caractéristiques du Produit (RCP) des traitements concomitants. Les patients doivent être traités par KEYTRUDA jusqu’à progression de la maladie ou toxicité inacceptable (et jusqu’à la durée maximale du traitement si spécifiée pour une indication). Des réponses atypiques (c’est-à-dire une augmentation initiale et transitoire de la taille de la tumeur ou l’apparition de nouvelles lésions de petite taille durant les premiers mois, suivies d’une régression de la tumeur) ont été observées. Chez les patients cliniquement stables présentant une progression initiale de la maladie, il est recommandé de poursuivre le traitement jusqu’à ce que la progression soit confirmée. Dans le traitement adjuvant du mélanome, KEYTRUDA doit être administré jusqu’à récidive de la maladie, toxicité inacceptable ou pendant une durée allant jusqu’à un an. Suspension ou arrêt définitif du traitement Aucune réduction de dose de KEYTRUDA n’est recommandée. KEYTRUDA doit être suspendu ou arrêté pour gérer les effets indésirables tels que décrit dans le tableau 1 du RCP. Populations particulières Personnes âgées Aucune adaptation posologique n’est nécessaire chez les patients âgés de ≥ 65 ans. Insuffisance rénale Aucune adaptation posologique n’est nécessaire pour les patients présentant une insuffisance rénale légère ou modérée. KEYTRUDA n’a pas été étudié chez les patients présentant une insuffisance rénale sévère Insuffisance hépatique Aucune adaptation posologique n’est nécessaire pour les patients présentant une insuffisance hépatique légère ou modérée. KEYTRUDA n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère Population pédiatrique La sécurité et l’efficacité de KEYTRUDA chez les enfants de moins de 18 ans n’ont pas été établies, sauf pour les patients pédiatriques atteints d’un mélanome ou d’un LHc. Contre-indications Hypersensibilité à la substance active ou à l’un des excipients suivants :L-histidine Chlorhydrate de L- histidine monohydraté Saccharose Polysorbate-80 (E433) . Mises en garde spéciales et précautions d’emploi Traçabilité Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés. Evaluation du statut PD-L1Lors de l’évaluation du statut PD-L1 de la tumeur, il est important de choisir une méthodologie robuste et validée pour minimiser les faux-négatifs et les faux-positifs. Effets indésirables à médiation immunitaire Des effets indésirables à médiation immunitaire, y compris des cas sévères et d’issue fatale, ont été rapportés chez des patients recevant pembrolizumab. La plupart des effets indésirables à médiation immunitaire apparus au cours du traitement par pembrolizumab ont été réversibles et pris en charge par une interruption de pembrolizumab, l’administration de corticostéroïdes et/ou des soins de support. Des effets indésirables à médiation immunitaire ont également été rapportés après la dernière administration de pembrolizumab. Des effets indésirables à médiation immunitaire affectant plus d’un système d’organes peuvent survenir simultanément. En cas de suspicion d’effets indésirables à médiation immunitaire, il convient de faire une évaluation appropriée pour confirmer l’étiologie ou éliminer les autres causes. En fonction de la sévérité de l’effet indésirable, pembrolizumab doit être suspendu et des corticostéroïdes doivent être administrés. En cas d’amélioration au Grade ≤ 1, une diminution progressive de la corticothérapie doit être initiée et poursuivie sur une période d’au moins 1 mois. Sur la base de données limitées issues des études cliniques chez les patients dont les effets indésirables à médiation immunitaire n’ont pu être contrôlés par des corticostéroïdes, l’administration d’autres immunosuppresseurs systémiques peut être envisagée. Si l’effet indésirable s’améliore jusqu’au Grade ≤ 1 et si la dose de corticostéroïdes est réduite à ≤ 10 mg de prednisone ou équivalent par jour, pembrolizumab peut être repris dans les 12 semaines suivant la dernière administration de KEYTRUDA. En cas d’effet indésirable à médiation immunitaire de Grade 3 récurrent et pour toute toxicité due à un effet indésirable à médiation immunitaire de Grade 4, pembrolizumab doit être arrêté définitivement, à l’exception des endocrinopathies contrôlées par traitement hormonal substitutif (Voir RCP pour plus de détails) Interactions avec d’autres médicaments et autres formes d’interactions Aucune étude pharmacocinétique formelle d’interaction médicamenteuse n’a été réalisée avec pembrolizumab. Pembrolizumab étant éliminé de la circulation par catabolisme, aucune interaction médicamenteuse métabolique n’est attendue. L’utilisation de corticostéroïdes ou d’immunosuppresseurs systémiques doit être évitée avant le début du traitement par pembrolizumab, du fait de leur possible interférence avec l’activité pharmacodynamique et l’efficacité de pembrolizumab. Néanmoins, les corticostéroïdes systémiques ou d’autres immunosuppresseurs peuvent être utilisés après l’instauration de pembrolizumab pour traiter des effets indésirables à médiation immunitaire. Les corticostéroïdes peuvent aussi être utilisés en prémédication quand pembrolizumab est utilisé en association à une chimiothérapie, en prophylaxie antiémétique et/ou pour atténuer les effets indésirables liés à la chimiothérapie. Fertilité, grossesse et allaitement Femmes en âge de procréer Les femmes en âge de procréer doivent utiliser une méthode efficace de contraception pendant le traitement par pembrolizumab et pendant au moins 4 mois après la dernière administration de pembrolizumab. Grossesse Il n’existe pas de données sur l’utilisation de pembrolizumab chez la femme enceinte. Aucune étude de reproduction n’a été conduite avec pembrolizumab ; cependant, dans des modèles murins de grossesse, le blocage de la voie de signalisation PD-L1 a montré une modification de la tolérance au foetus et a conduit à une augmentation des pertes fœtales. Sur la base du mécanisme d’action, ces résultats indiquent que l’administration de pembrolizumab pendant la grossesse expose à un risque d’effet nocif sur le foetus, incluant des taux plus élevés d’avortement ou d’enfants mort-nés. Il est connu que les immunoglobulines humaines G4 (IgG4) passent la barrière placentaire ; par conséquent pembrolizumab, étant une IgG4, a la possibilité d’être transmis de la mère au foetus en développement. Pembrolizumab ne doit pas être utilisé pendant la grossesse à moins que l’état clinique de la femme ne nécessite un traitement par pembrolizumab. Allaitement On ne sait pas si pembrolizumab est excrété dans le lait maternel. Les anticorps étant connus pour être secrétés dans le lait maternel, un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d’interrompre l’allaitement soit d’interrompre pembrolizumab, en prenant en compte le bénéfice de l’allaitement pour l’enfant et le bénéfice du traitement par pembrolizumab pour la femme. Fertilité Aucune donnée clinique n’est disponible concernant les effets possibles de pembrolizumab sur la fertilité. Les études de toxicité à dose répétée de 1 mois et 6 mois menées sur des singes n’ont pas montré d’effets notables sur les organes reproducteurs mâles et femelles. Effets sur l’aptitude à conduire des véhicules et à utiliser des machines Pembrolizumab a une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines. Chez certains patients, des étourdissements et de la fatigue ont été rapportés après administration de pembrolizumab Effets indésirables Résumé du profil de sécurité Pembrolizumab est le plus fréquemment associé à des effets indésirables à médiation immunitaire. La plupart d’entre eux, y compris les réactions sévères, se sont résolus après initiation d’un traitement médical approprié ou arrêt de pembrolizumab (voir « Description d’une sélection d’effets indésirables » ci-dessous). Les fréquences mentionnées ci-dessous et dans le tableau 2 du RCP sont basées sur tous les effets indésirables rapportés, quelle que soit l’évaluation de la causalité par l’investigateur. Pembrolizumab en monothérapie La sécurité de pembrolizumab en monothérapie a été évaluée dans des études cliniques chez 7 631 patients dans différents types de tumeurs et avec quatre doses (2 mg/kg de poids corporel toutes les 3 semaines, 200 mg toutes les 3 semaines ou 10 mg/kg de poids corporel toutes les 2 ou 3 semaines). Dans cette population de patients, la durée d’observation médiane était de 8,5 mois (de 1 jour à 39 mois) et les effets indésirables les plus fréquents avec pembrolizumab étaient : fatigue (31 %), diarrhée (22 %) et nausée (20 %). La majorité des effets indésirables rapportés en monothérapie étaient d’une sévérité de Grades 1 ou 2. Les effets indésirables les plus graves étaient des effets indésirables à médiation immunitaire et des réactions sévères liées à la perfusion. Les incidences des effets indésirables à médiation immunitaire étaient de 37 % tous Grades, et 9 % pour les Grades 3-5 avec le pembrolizumab en monothérapie au stade adjuvant, et 25 % tous Grades, et 6 % pour les Grades 3-5 au stade métastatique. Aucun nouvel effet indésirable à médiation immunitaire n’a été identifié au stade adjuvant. Pembrolizumab en association à une chimiothérapie Lorsque pembrolizumab est administré en association, reportez-vous au RCP des médicaments respectifs du traitement en association avant l’initiation du traitement. La sécurité de pembrolizumab en association à une chimiothérapie a été évaluée dans des études cliniques chez 5 183 patients dans différents types de tumeurs recevant 200 mg, 2 mg/kg de poids corporel ou 10 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines. Dans cette population de patients, les effets indésirables les plus fréquents étaient : anémie (52 %), nausées (52 %), fatigue (35 %), diarrhée (33 %), constipation (32 %), vomissements (28 %), diminution de l’appétit (28 %), diminution du nombre de neutrophiles (27 %) et neutropénie (25 %). Les incidences des effets indésirables de Grades 3-5 chez les patients avec un CBNPC étaient de 69 % pour le traitement par pembrolizumab en association et de 61 % pour la chimiothérapie seule, chez les patients avec un CETEC étaient de 85 % pour le traitement par pembrolizumab en association et de 84 % pour la chimiothérapie avec cétuximab, chez les patients atteints d’un cancer de l’oesophage étaient de 86 % pour le traitement par pembrolizumab en association et de 83 % pour la chimiothérapie seule, chez les patients atteints d’un CSTN étaient de 80 % pour le traitement par pembrolizumab en association et de 77 % pour la chimiothérapie seule, chez les patientes atteintes d’un cancer du col de l’utérus étaient de 82 % pour le traitement par pembrolizumab en association et de 75 % pour la chimiothérapie, avec ou sans bevacizumab, chez les patients atteints d’un cancer gastrique étaient de 74 % pour le traitement par pembrolizumab en association (chimiothérapie avec ou sans trastuzumab) et de 68 % pour la chimiothérapie avec ou sans trastuzumab, et chez les patients atteints d’un carcinome des voies biliaires étaient de 85 % pour le traitement par pembrolizumab en association et 84 % pour la chimiothérapie seule. Pembrolizumab en association à un inhibiteur de tyrosine kinase (ITK) Lorsque pembrolizumab est administré en association à l’axitinib ou au lenvatinib, reportez-vous au RCP de l’axitinib ou du lenvatinib avant l’initiation du traitement. Pour des informations complémentaires sur la sécurité du lenvatinib pour le carcinome à cellules rénales (CCR) avancé, voir le RCP de Kisplyx et pour le cancer de l’endomètre (CE) avancé, voir le RCP de Lenvima. Pour des informations complémentaires sur la sécurité de l’axitinib en cas d’élévation des enzymes hépatiques. La sécurité de pembrolizumab en association à l’axitinib ou au lenvatinib dans le CCR avancé, et en association au lenvatinib dans le CE avancé, a été évaluée chez un total de 1 456 patients atteints d’un CCR avancé ou d’un CE avancé recevant 200 mg de pembrolizumab toutes les 3 semaines avec soit 5 mg d’axitinib deux fois par jour, soit 20 mg de lenvatinib une fois par jour dans les études cliniques, selon le cas. Dans ces populations de patients, les effets indésirables les plus fréquents étaient : diarrhée (58 %), hypertension (54 %), hypothyroïdie (46 %), fatigue (41 %), diminution de l’appétit (40 %), nausées (40 %), arthralgie (30 %), vomissements (28 %), perte de poids (28 %), dysphonie (28 %), douleurs abdominales (28 %), protéinurie (27 %), syndrome main-pied (26 %), éruption cutanée (26 %), stomatite (25 %), constipation (25 %), douleurs musculosquelettiques (23 %), céphalées (23 %) et toux (21 %). La fréquence des effets indésirables de Grade 3-5 chez les patients atteints de CCR était de 80 % pour le pembrolizumab en association à l’axitinib ou au lenvatinib et de 71 % pour le sunitinib seul. Chez les patients atteints de CE, la fréquence des effets indésirables de Grade 3-5 était de 89 % pour le pembrolizumab en association au lenvatinib et de 73 % pour la chimiothérapie seule.
Pour tout complément d’information, veuillez consulter le Résumé des Caractéristiques du Produit (RCP)
Réferences:
- KEYTRUDA RCP.
- Bocanegra A, Fernandez-Hinojal G, Zuazo-Ibarra M, Arasanz H, Garcia-Granda MJ, Hernandez C, et al. PD-L1 expression in systemic immune cell populations as a potential predictive biomarker of responses to PD-L1/PD-1 blockade therapy in lung cancer. International journal of molecular sciences. 2019;20(7):1631.
- Dako AP (FDA-approved for in vitro diagnostic use ). PD-L1 IHC 22C3 pharmDx Interpretation Manual-–Head and Neck Squamous Cell Carcinoma (HNSCC. Carpinteria, CA. 2019.
- de Ruiter EJ, Mulder FJ, Koomen BM, Speel EJ, van den Hout MF, de Roest RH, et al. Comparison of three PD-L1 immunohistochemical assays in head and neck squamous cell carcinoma (HNSCC). ModernPathology. 2021;34(6):1125-32.
- Agilent Technologies, Inc. PD-L1 IHC 22C3 pharmDx Interpretation Manual-Non-small Cell Lung Cancer (NSCLC).
- Frančina M, Mikuš M, Mamić M, et al. Evaluation of PD-L1 Expression in Colorectal Carcinomas by Comparing Scoring Methods and Their Significance in Relation to Clinicopathologic Parameters. Diagnostics (Basel). 2024;14(10):1007.
DZ-KEY-00261. Date d’expiration : 23.Décembre.2025